
Информация для специалистов || Патогенез
ПАТОГЕНЕЗ
В связи с тем, что вирусы являются патогенами внутриклеточными, каждый представитель мира вирусов имеет тропность к определенному типу клеток. Тропизм вируса определяется наличием на клетке - мишени рецептора для данного вируса, а также возможность генома вируса встроиться в геном клетки. Рецепция в свою очередь определяет: а) конкретный вирус взаимодействует только с определенными рецепторами, б) на клетке могут быть рецепторы для различных типов вирусов и в) рецепторы для определенного вируса могут быть на клетках различных типов. Рецепторную функцию выполняют различные структуры (лиганды): белки, липиды, углеводные компоненты белков и липидов. Эти лиганды локализованы на плазматической мембране выполняют важнейшие функции жизнеобеспечения клетки - проникновение в нее гормонов, питательных веществ, факторов роста и регуляции и т.п. Вирус использует эти рецепторы в своих целях.
Рецепторы, независимо от их биохимического строения, имеют общую структурную характеристику, а именно состоят из участка, расположенного вне клетки, участка, локализованного внутримембранно и участка, погруженного в цитоплазму.
Рецептором для ВИЧ является дифференцировочный антиген CD4 - (СD - аббревиатура от Сell Differention antigen), а также неспецифические, независящие от наличия CD4 компоненты. CD4 -гликопротеид с молекулярной массой 55 000, по своему строению имеющий гомологии с определенными участками иммуноглобулинов. Аналогичные гомологии имеет и белок вируса gp 120, что и определяет тропность ВИЧ. Расположенный на мембране иммунокомпетентных клеток Т-хелперов и Т- индукторов, рецептор CD4 выполняет функцию распознавания (в комплексе с белками HLA II класса) антигенов. Фиксация вируса через gp120 ВИЧ-1 (или gp 105 в случае инфицирования ВИЧ-2) с мембранным рецептором CD4 клетки хозяина (рис. 5) блокирует основную функцию этих иммунокомпетентных клеток - восприятие сигналов от антигенпрезентирующих клеток. Последующая за рецепцией репликация вируса ведет к гибели клеток, выпадению функции, ими выполняемой - развитие иммунодефицита.
Рис. 5. Этапы взаимодействия ВИЧ с CD4-клеткой (Т-хелпером)
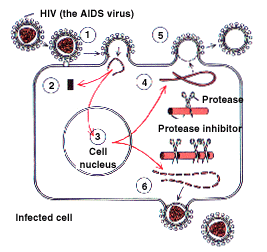
Обозначения:
-
Связывание ВИЧ с рецептором CD4
-
РНК ВИЧ переписывается на ДНК с помощью обратной транскриптазы с превращением в ДНК-провирус
-
Интеграция провируса с помощью интегразы в ядро CD4-клетки, синтез геномной РНК для новых вирионов
-
Трансляция вирусных белков и расщепление протеазой вирусных полипептидов
-
Сборка новых вирионов
-
Выход новых вирионов из клетки-мишени
В организме человека имеется целый ряд иммунокомпетентных, соматических и ряда других клеток, имеющих рецепторы для ВИЧ (табл. 16) и наблюдается цитопатический эффект во многих из них в случае проникновения вируса.
Таблица 16. Клетки-мишени для ВИЧ
|
Тип клеток |
Наличие CD4 |
Цитопатический эффект вируса |
|
Система крови- |
||
|
- CD4+ лимфоциты |
+ |
+ |
|
- CD8+ лимфоциты |
+ |
+ |
|
- Дендритные клетки |
+ |
- |
|
- Моноциты/макрофаги |
+ |
- |
|
- Эозинофилы |
+ |
? |
|
- Мегакариоциты |
+ |
+ |
|
- Тимоциты-предшественники |
||
|
CD4+ и CD8+ клеток |
+ |
+ |
|
- В-лимфоциты |
на некоторых |
некоторые линии |
|
Нервная система: |
||
|
- Нейроны |
+ |
|
|
- Микроглия |
+ |
|
|
- Астроциты |
? |
? |
|
Клетки хорионтрофобласта |
||
|
плаценты |
+ |
|
|
Спетрматозоиды |
+ |
- |
Помимо основного рецептора для ВИЧ-1 – CD4 имеется еще ряд корецепторов, в частности, хемокиновые рецепторы, необходимые для проникновения ВИЧ в клетку.
Хемокины – полипептиды, вызывающие движение клеток в определенной направленности. У человека выделено около 40 отдельных подобных белков, их подразделили на альфа- и бета-хемокины. В лаборатории Галло в 1995 году был выделен хемокин из CD8-лимфоцитов и два белка из макрофагов. Эти три хемокина блокируют инфицирование CD+ мононуклеаров макрофаготропными, но не лимфотропными вариантами ВИЧ-1.
В 1996 году Бергер открыл корецептор для ВИЧ, сегодня получивший название СХСR4. Этот рецептор вместе с CD4 опосредует инфицирование СО-вариантами ВИЧ 1, адаптированных к росту в Т-хелперах, но не в макрофагах. В 1996 году открыт еще один корецептор для ВИЧ 1 – ССR5. На сегодня известен ряд хемокиновых рецепторов (табл. 17).
Таблица 17. Хемокиновые рецепторы человека
|
Клетки |
Рецепторы |
Хемокиновые лиганды |
|
Нейтрофилы, моноциты |
CXCR 1 |
ИЛ-8 |
|
Нейтрофилы, моноциты, базофилы |
СХСR 2 |
ИЛ-8, белок, активирующий |
|
нейтрофилы |
|
|
|
Нормальные и активированные Т- |
CXCR 3 |
ИФ-10 |
|
клетки, В-клетки, моноциты, макро- фаги, гранулоциты, дендритные клетки |
CXCR 4 |
Фактор-1 стромальных клеток |
|
Моноциты, базофилы, активи рованные Т- клетки |
CCR1 |
Регулятор и активатор нормальных Т-клеток и др. |
|
Моноциты, базофилы, активированные Т- клетки |
CCR1 |
Регулятор и активатор нормальных Т-клеток и др. |
|
- II - |
ССR2 |
Белок, вызывающий хемотаксис моноцитов 1, 2, 3, 4 |
|
Эозинофилы |
ССR3 |
Белок, вызывающий хемотаксис эоинофилов, белок, вызывающий хемотаксис моноцитов-3,4, регу- лятор и активатор нормальных Т-клеток |
|
Моноциты, активированные Т-клетки |
ССR4 |
Регулятор и активатор нормальных Т-клеток и др. |
|
Активированные моноциты и Т-клетки, дендритные клетки |
CCR5 |
- II - |
Таким образом, выделены белки - хемокины, блокирующие проникновение ВИЧ в макрофаги с антигеном CD4 и белки - корецепторы, способствующие инфицированию. При этом корецепторы – это рецепторы для хемокинов, но их использует ВИЧ в качестве рецептора, с помощью которого проникает внутрь клетки. Некоторые хемокиновые рецепторы необходимы для проникновения ВИЧ в клетку.
Было установлено, что длительно контактирующие с ВИЧ-инфицированными и при этом не заражающиеся имеют мутации в рецепторе CCR5, поэтому мононуклеары у них оказались высокорезистентными к ВИЧ.
Проникнув в СD4+ клетки ВИЧ сразу же начинает репликацию, при этом, чем активнее CD4+ клетки, тем выше процесс репродукции вируса. А отсюда вытекает, что все регуляторы, активирующие СD4+клетки, обеспечивают увеличение репликации вируса. К подобным регуляторам относятся фактор некроза опухолей (ФНО), фактор, стимулирующий колонии гранулоцитов/макрофагов, интерлейкин-6 (ИЛ-6). К негативным регуляторам, тормозящим репликацию вируса, относятся интерферон (ИФ), трансформирующий фактор роста. Так, установлено, что ФНО-a активирует
транскрипцию провирусной ДНК ВИЧ 1 в хронически инфицированных Т-клетках и макрофагах. Моноциты продуцируют ФНО-a , они не только индуцируют экспрессию ВИЧ-инфицированными клетками, но они также стимулируют активацию латентного провируса ВИЧ. Синхронно с ФНО-a действуют ИЛ-6 и фактор, стимулирующий колонии гранулоцитов/макрофагов.
Морфо-функциональные изменения в моноцитах/макрофагах оказывают разнонаправленное действие не только на Т-лимфоциты, но и естественные киллеры - главные клетки противоопухолевой защиты. Активность последних по мере прогрессирования заболевания неуклонно снижается. Уже дефицит ИЛ-2 и гамма-интерферона даже при нормальном количестве NK-клеток ведет к снижению функциональной активности их у больных ВИЧ-инфекцией (Ковальчук Л.В., Чередеев А.Н.,1991), тем более при дефиците. Хотя NK-клетки принимают участие в противоопухолевой защите, при ВИЧ-инфекции ведущим рассматривается иной механизм развития онкопатологии (рис. 6).
Рис. 6. Механизм
индукции опухолевого роста при ВИЧ-инфекции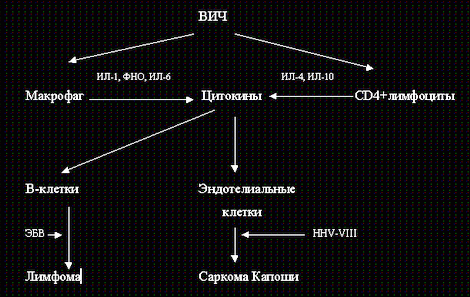
Выделено два типа CD4+клеток: Т-хелперы –1 и Т-хелперы-2. Т-хелперы – 1 продуцируют цитокины, стимулирующие клеточный иммунитет, а Т-хелперы-2 – цитоктины, усиливающие антителогенез. Соотношение Т-хелперы-1 и Т-хелперы-2 взвешенно и конкурентно, суперэкспрессия цитокинов одного типа клеток ведет к супрессии другого. У больных ВИЧ-инфекцией идет угнетение Т-хелперов-1, чем обеспечивается и вирусная патология и онкогенез.
Жизненный цикл ВИЧ после проникновения в организм носит последовательный характер: связывание вириона с поверхностью клетки, слияние мембран вириона и клетки, проникновение вируса внутрь клетки, высвобождение нуклеотида и геномной РНК вируса, интеграция генома вируса в геном инфицированной клетки, латентная фаза, фаза активации транскрипции с ДНК провируса и последующая транскрипция белков вируса, наработка всех компонентов вируса с формированием новых вирионов и их высвобождением из клетки, влекущим за собой гибель клетки-мишени.
Сродство вирусного мембранного гликопротеида gp120 (gp105 в случае ВИЧ-2) к клеточному рецептору CD4 определяет высокую степень избирательного поражения клеточных структур. Поэтому в патологический процесс вовлекаются в первую очередь и в большей степени CD4+ лимфоциты, моноциты крови, макрофаги тканей, дендритные клетки крови, лимфатических узлов, селезенки, кожи, альвеолярных и интерстициальных макрофагов легких, микроглия и другие клетки нервной системы, имеющие CD4 - рецепторы. Так же поражаются В- и О-лимфоциты, ретикулярные клетки, эпителиальные клетки кишечника, клетки Лангерганса. Причем последние инфицируются даже легче, чем CD4+ лимфоциты. Именно клеткам Лангерганса придается большое значение в распространении ВИЧ по организму, ибо в них вирус сохраняется длительное время, иногда годы.
Наличие CD4 рецептора на многих и не только иммунокомпетентных клетках, возможность поражать и клетки, не имеющие этого рецептора, определяют политропность ВИЧ и полиморфизм клинической картины. Степень поражения тех или иных, содержащих СD4 рецепторы клеток зависит от плотности этих рецепторов на мембране клеток. Наиболее высока плотность на Т-хелперной субпопуляции лимфоцитов, что и определяет во многом патогенез болезни. Но степень поражения клеток-мишеней вирусом зависит также и от возможности репликации вируса в том или ином виде клеток. Очевидно, осуществляется репликация в основном в лимфоцитах с CD4+ фенотипом и моноцитах/макрофагах.
Если на CD4+ лимфоциты вирус оказывает цитопатическое действие с лизисом клетки или слиянием в синцитий, то в моноцитах/макрофагах ВИЧ реплицируется с умеренной интенсивностью, вирионы оформляются в округлые частицы еще в цитоплазме клетки и по выходе из нее не оказывают цитонекротического действия. Тем не менее, моноциты/ макрофаги претерпевают значительные ультраструктурные изменения. Проведенное нами изучение динамики сывороточных монокинов подтверждает активное участие в патогенезе ВИЧ-инфекции моноцитов/ макрофагов, основных продуцентов фактора некроза опухолей и интерлейкина-1b (выполняющих роль передатчика Т-лимфоцитам антигенспецифических сигналов, необходимых для их активации в иммунном ответе). Увеличение продукции некоторых цитокинов способствует экспрессии вируса. Так, исследования Н.М. Калининой (1996) показали резкое увеличение провоспалительных цитокинов ФНО-a (до 428 при норме 38,9 пг/мл у взрослых и до 1363 у детей при норме 108,8 пг/мл в стадии IIIB) и ИЛ-1b . С гиперпродукцией ФНО-a , ИЛ-1b и ИЛ-6 связывают развитие при ВИЧ-инфекции лихорадки, анемии, диареи, кахексии, патологических изменений на коже и слизистых при саркоме Капоши, церебральных симптомов. ФНО-a при этом оказывает прямое цитопатическое действие на инфицированные ВИЧ Т-хелперы. В то же время было установлено, что ВИЧ ингибирует продукцию ИЛ-2 и ИФ-g , синтезируемым Т-хелперами первого типа и не ингибирует функцию Т-хелперов второго типа. Следовательно, играя важную роль в регуляции синтеза цитокинов, ВИЧ за счет переключения иммунного ответа с Т-хелперов первого типа на Т-хелперы второго типа, стимулирует гуморальное звено иммунитета.
После инфицирования клетки вирусом происходит соединение вирусной оболочки с помощью белка gp41 с мембраной клетки. Помимо того вирусный белок gp41 обеспечивает слияние мембран соседних клеток между собой с образованием одной многоядерной клетки - синцития. При этом слияние может быть как зараженных клеток между собой, так и зараженных с незараженными. Но синцитий в основном индуцируют вирусы, выделенные от больных с клиническими проявлениям ВИЧ-инфекции и не образуют выделенные от инфицированных, не имеющих клинических проявлений.
С момента интеграции генома вируса в геном клетки начинается стадия латентной инфекции. В этот период вирус находится в клетке в виде интегрированного в геном ДНК - провируса,. Раньше полагали, что в этот период нет процессов транскрипции и трансляции с вирусных генов, а поэтому и экспрессии вирусных белков, нет иммунного ответа на вирус как в виде специфических иммуноглобулинов, так и сенсибилизированных лимфоцитов. Но последние исследования показали, что сразу после проникновения вируса в клетку начинается и трансляция и транскрипция.
В этот период инфекционного процесса, когда возбудитель находится в геноме клетки в виде ДНК-провируса, его можно обнаружить лишь такими методами, как полимеразная цепная реакция (ПЦР), гибридизация нуклеиновых кислот со специфическими зондами. ПЦР позволяет обнаружить вирус при наличии одной инфицированной из 5 тыс. клеток.
Сохранение инфекционного процесса в латентной форме обеспечивается посредством элемента негативной регуляции (NRE), идентифицированного в участке длинного концевого повтора ВИЧ. Делеция NRE приводит к усилению экспрессии вирусного генома. К NRE имеет сродство вирусный белок nef. Тем не менее, один из штаммов ВИЧ-1, несмотря на присутствие nef является высоковирулентным, что объясняется наличием двух мутантов nef .
Состояние латентной инфекции без клинических признаков болезни может длиться от 2 до 11 лет. Активация длинных концевых повторов и экспрессия генов ВИЧ, кодирующих белки вируса, знаменует собой манифестацию болезни.
В расшифровке патогенеза ВИЧ-инфекции многие вопросы решены, установлены основные этапы развития процесса в клетке после инфицирования ее вирусом. Выявлен ряд факторов, активирующих экспрессию генов ВИЧ (табл. 18). К ним относятся факторы, активирующие Т-лимфоциты: специфические антигены (например, вирусы группы Herpes), неспецифические антигены (например, митогены типа фитогемаглютинина), цитокины (например, фактор некроза опухолей, некоторые интерлейкины, гамма-интерферон), бактериальные иммуномодуляторы (например, монофосфат липида из сальмонелл). К активаторам экспрессии ВИЧ относятся глюкокортикостероидные гормоны, в частности дексаметазон и гидрокортизон, ультрафиолетовое облучение, перекись водорода, свободные кислородные радикалы. Активирует инфекционный процесс беременность, более прогрессирующее течение ВИЧ-инфекции имеют психически неуравновешенные лица, дезадаптированные по сравнению с людьми, имеющими уравновешенную психику.
Таблица 18. Ко-факторы ВИЧ-инфекции
|
I.Фаза интегрирования |
|
- травмы гениталий |
|
- болезни, передающиеся половым путем |
|
- вирусная инфекция (простой герпес, цитомегаловирусная инфекция и др.) |
|
II. Манифестация острой фазы (у 15-25% ВИЧ-инфицированных) |
|
- вирусная инфекция группы Herpes |
|
III. Фаза перехода из латентной в активную |
|
- генетические факторы (ассоциация с HLA) |
|
- возраст |
|
- ультрафиолетовые лучи и др. факторы |
Довольно сложен и до конца не выяснен механизм репликации вируса в инфицированной клетке. Известно, что в цитоплазме информация с вирусной РНК посредством обратной транскриптазы (ревертазы) переписывается на ДНК клетки, первоначально образуется однонитевая структура, затем та же обратная транскриптаза обеспечивает образование второй нити, а линейная промежуточная форма ДНК вируса транспортируется в ядро, где с помощью фермента интегразы интегрирует с ДНК клетки, превращаясь в ДНК-провирус.
Исключительно важный этап патогенеза болезни - сборка вирусных частиц и выход нового потомства вируса из инфицированной клетки. Сборка происходит на плазматической мембране лимфоцита, куда поступают все компоненты вирусной частицы, в том числе и белки-предшественники. Завершается процесс почкованием вирусных частиц с клеточной поверхности. Отличительной чертой ВИЧ является взрывной характер процессов активации транскрипции, синтеза белков-предшественников, сборки вирионов и их почкования: за 5 мин одна лимфоцитарная клетка может образовать до 5000 вирусных частиц.
Ключевой вопрос патогенеза ВИЧ-инфекции - механизм иммунного повреждения. Как установлено, в составе белков gp120, главного комплекса гистосовместимости (HLA) класса II и CD4-рецепторов имеются сходные участки, что определяет перекресное реагирование образующихся к ВИЧ антител с этими структурами. Например, происходит блокада кооперации CD4+ лимфоцитов и HLA II. Антитела к gp120 ВИЧ реагируют с CD4, обусловливая неадекватную стимуляцию CD4+ клеток. На всех ядросодержащих клетках имеются антигены HLA I, вирус же нарушает синтез этих антигенов, участвующих в распознавании CD8+ лимфоцитами (Т-супрессорами) зараженных вирусом клеток, что тормозит процесс лизиса инфицированных клеток.
Взаимосвязь жизненного цикла вируса и иммунных сдвигов в начальной фазе ВИЧ-инфекции изучена в эксперименте при заражении SIV обезьян, полученные данные экстраполированы на человека. Как показали исследования, вирус первоначально локализуется в лимфоидной системе, вирусная экспрессия определяет клинику ранней фазы болезни. Пик экспрессии вируса в клетках лимфоидной ткани предшествует накоплению его в плазме. Появление вирусспецифических цитотоксических Т-лимфоцитов (CTL) совпадает со временем окончания экспрессии вируса в лимфатических узлах. Продукция комплемент (С) - связывающих антител облегчает переход вируса в сеть дендритных клеток герминальных центров лимфатических узлов. Повреждающее действие CTL и продукция копмлементсвязывающих антител оказывают основное патогенное действие в результате виремии. Образование нейтрализующих антител обеспечивает переход острой фазы болезни в хроническую.
Как показали исследования, проведенные в нашей лаборатории Ю.А.Митиным, в ходе развития инфекционного процесса на всех фазах болезни наблюдаются морфологические изменения лимфоцитов: увеличивается содержание сегментированных лимфоцитов, усиливается деградация нуклеотидов, снижаются биосинтетические процессы и нарушается организация надмолекулярного комплекса ДНК. Острая фаза болезни приводит к усилению митотической и предмитотической активности лимфоцитов, на стадии II B растет митотическая активность моноцитов периферической крови. Патологический процесс сопровождается усилением перекисного окисления липидов в лимфоцитах. Снижается активность ключевого фермента анаэробного окисления - лактатдегидрогеназы, характерно повышение активности сукцинатдегидрогеназы, снижение активности альфа-глицерофосфатдегидро-геназы.
В моноцитарных клетках больных изменения функциональной активности проявляются повышением напряженности процессов анаэробного окисления на ранних стадиях инфицирования с одновременным снижением интенсивности цикла трикарбоновых кислот. Прогрессирование патологического процесса ведет к стойкому снижению активности сукцинатдегидрогеназы и лактатдегидрогеназы, характеризуя угнетение функциональной активности моноцитов.
Активация пентозного цикла, усиление процесса распада пуриновых оснований приводят к активации прооксидантных НАД-систем (НАДФ и ксантиноксидазы), не компенсируемый при прогрессировании патологического процесса усилением функции антиоксидантных систем (супероксиддисмутаза, глутатион) и обусловливая рост активности трипсиноподобных эндонуклеаз с развитием деградации ядерного хроматина. Все это определяет несбалансированный рост и преждевременную гибель клеток.
Иммунопатогенетически ВИЧ-инфекция проявляется дефицитом Т- и В-звеньев иммунной системы, дефицитом комплемента, фагоцитов, снижением функции неспецифических факторов защиты. В результате идет формирование анергии с проявлениями аллергического, аутоаллергического и иммунокомплексного патологического процесса. Так, наши исследования показали, что уже во II стадии ВИЧ-инфекция характеризуется снижением абсолютного числа лейкоцитов за счет CD3+, CD4+, p24+ и gp120+ лимфоцитов, повышением уровня естественных киллеров (NК-клетки), нарастанием РТМЛ с конкавалином А и фитогемагглютинином. Изменения гуморального звена иммунитета проявляются нарастанием IgG+ В-лимфоцитов и 4-5 кратным ростом уровня сывороточных IgE.
Как показали исследования, проведенные в нашей лаборатории, количественные и функциональные сдвиги в показателях иммунной системы индуцируют изменения структурной организации ее с установлением новых и изменением силы и направленности ранее имевшихся коррелятивных связей: уже на стадии II Б возникает положительная коррелятивная связь между содержанием CD8+ лимфоцитов и числом иммунокомпетентных клеток, имеющих на поверхностных мембранах антиген ВИЧ р24. В стадии III A устанавливается тесная положительная коррелятивная связь между числом NK-клеток, индексом CD4/ CD8 и содержанием клеток, экспрессирующих антигены ВИЧ р24 и gp120. В стадии III Б и III B возрастает число клеток, экспрессирующих антигены p24 и gp 120, устанавливается высокая отрицательная коррелятивная связь между числом CD3+ лимфоцитов и содержанием клеток, экспрессирующих антигены р24 и gp120, между числом NK-клеток и числом клеток, экспрессирующих антиген р24.
Для изменений В-звена иммунитета характерны активация В-лимфоцитов, изменение их метаболизма сопровождается уменьшением времени полуобмена иммуноглобулиновых рецепторов с повышением в крови концентрации клеточных рецепторов (Р-белки). Суммарная концентрация сывороточных иммуноглобулинов растет, но выявляется диспропорция уровней подклассов иммуноглобулинов. Так, содержание IgG1 и IgG3 у больных увеличивается, а концентрация IgG2 и IgG4 существенно уменьшается. Очевидно, повышение уровня IgG2 связано с высокой восприимчивостью больных к стафилококкам, пневмококкам, палочке инфлюэнцы. Несмотря на гипергаммаглобулинемию, увеличение количества циркулирующих в крови В-лимфоцитов, их функциональная активность на митогены (например, на митоген лаконоса) остается относительно сниженной, что позволяет В-систему иммунитета у ВИЧ-больных расценивать как супрессированную. К тому же, как показывают наши наблюдения и данные литературы, количество В-клеток в периферической крови больных в стадии СПИД может быть снижено в три и более раз.
Многочисленными исследованиями показано, что взаимодействие gp 120 ВИЧ-1 с мембраной CD4+ лимфоцитов определяет не только отрицательные реакции инфицированных клеток, но и приводит к программированной клеточной гибели - апоптозу зрелых CD4+ лимфоцитов или CD34+ гемопоэтических клеток-предшественников даже при отсутствии их инфицирования вирусом.
Специфический иммунный ответ при ВИЧ-инфекции имеет ряд особенностей. Локализуясь в иммуноците, вирус проникает в органы и ткани, пребывая вне доступности для иммунной системы, чем определяется персистенция в клетках как вируса, так и его компонентов - белков, нуклеокапсидов, нуклеиновой кислоты. Активация инфекционного процесса ведет к гуморальному иммунному ответу с образованием антител. Но в связи с наличием в составе вирусного белка gp120, HLA класса II и CD4-рецептора лимфоцита сходных участков, образующиеся антитела перекресно реагируют с ними, чем обусловливают нарушение кооперативных взаимодействий в функционировании иммунной системы. Все это определяет формирование аутоаллергических реакций. Именно поэтому в ходе развития инфекционного процесса формируется гиперчувствительность замедленного и немедленного типов, преимущественно к антигенам ВИЧ, в частности gр41 и gp120. Так, Л.Монтанье (1996) считает, что в патогенезе СПИД основное место принадлежит аутоиммунному процессу. В пользу этого говорят исследования характера иммунных реакций у больных: наличие аутоантител и аллоантител к лимфоцитам, формирование аллергических реакций цитотоксического типа, накопление циркулирующих иммунных комплексов, наличие комплементопосредованной цитотоксичности, активация антителозависимой клеточной цитотоксичности. Кроме того, при сильном антительном ответе возможность развития аутоиммунных процессов обусловлена мимикрией структуры gp120 под структуру иммуноглобулинов (Atlan H. et al.,1993). В пользу наличия аутоиммунных процессов в патогенезе ВИЧ-инфекции говорят и клинические проявления болезни, в частности, часто сопутствующие болезни такие типичные аутоаллергические варианты патологии, как спондилоартропатии, системные ревматоидные синдромы: волчаночноподобный, синдром Шегрена, полимиозит, некротизирующий васкулит. Возможный вариант развития аутоиммунного синдрома у больных ВИЧ/СПИД представлен на рис. 7.
Рис. 7. Возможные механизмы аутоаллергических нарушений при ВИЧ-инфекции
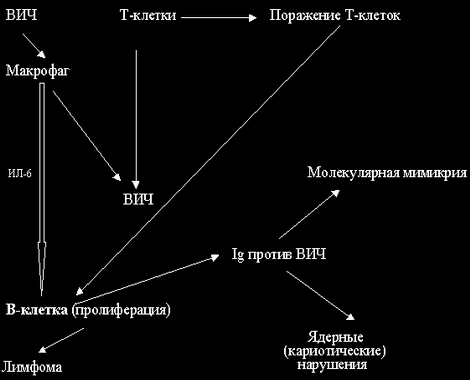
Исследования, проведенные Ю.А.Митиным (1997) показали, что у половины больных (52,4%) развиваются манифестные формы аллергии, преимущественно в виде ринита (46,1%), лекарственной аллергии (31,0%), крапивницы (17,2%), атопического дерматита (16,1%), ангионевротического отека (2,3%) с повышением у них уровня специфических IgE к бытовым, эпидермальным, пыльцевым и пищевым аллергенам с положительной коррелятивной связью между общим уровнем IgE и содержанием СD8+ лимфоцитов. В целом аллергическая настроенность характеризуется ранним началом от момента заболевания, развитием полисенсибилизации.
Формирование иммунодефицита при ВИЧ-инфекции не ограничивается только поражением лимфоцитов с CD4-фенотипом. Так, нарушение синтеза белков HLA I ведет к ингибированию функции лимфоцитов с CD8-фенотипа, т.е. Т-супрессоров. Белок вируса р15 оказывает супрессивное действие на продукцию Т-клетками ИЛ-2 и интерферона-g . Известно, что для дифференцировки Т-эффекторов из Т-предшественников необходимы ИЛ-2, гамма-интерферон и ИЛ-6. А с продукцией ИЛ-2 и других цитокинов тесно связана функция цитотоксических Т-лимфоцитов, ответственных за противовирусную и противоопухолевую защиту организма. Все это патогенетически определяет характерную для ВИЧ-инфекции СПИД-ассоциированную вирусную и онкопатологию.
Наряду с поражением иммунной системы в патологический процесс вовлекается и кроветворная ткань. Для заболевания характерны лейкопения, анемия, тромбоцитопения. Угнетается функциональная активность гранулоцитов. Экспериментальные исследования с заражением ВИЧ-подобным ретровирусом LP-BM5 через 3 месяца в костном мозге выявлена значительная цитопения, снижение содержания эрелых клеток, угнетение гранулоцитопоэза (Magnal M.et al., 1993). У больных ВИЧ-инфекцией в костном мозге резко снижено содержание колониеобразующих единиц гранулоцитов, макрофагов, мегакариоцитов. Однако дисфункция стволовых клеток не связана с их прямым инфицированием, ибо не обнаружено присутствия ВИЧ в кроветворных клетках при изучении методом гибридизации in situ и иммуногистохимическим исследованием гемопоэтических колоний, а также с помощью полимеразной цепной реакции. Пока нет однозначного мнения о причинах ингибиции пролиферативной активности стволовых клеток, хотя и установлено, что различные изоляты ВИЧ, воздействуя на CD34+ клетки, угнетают их функциональную активность. Также установлено, что подавление кроветворения в результате действия ВИЧ связано с усилением продукции костномозговыми моноцитами/макрофагами фактора некроза опухолей.В целом патогенез ВИЧ-инфекции схематично представлен на рис. 8. Причем, накопленные в последние годы данные позволили изменить имевшееся ранее представление о патогенезе ВИЧ-инфекции.
Рис. 8. Схема патогенеза ВИЧ-инфекции
|
|
<----- |
ВИЧ |
-----> |
|
|
Моноцит/ макрофаг
|
|
СD4+лимфоцит
|
|
|
Латентная
моноцитарно/макрофагальная инфекция |
|
Латентная
Т4-клеточная инфекция |
|
Ко-факторы (инфекция,особенно CMV-инфекция, ультрафиолетовое облучение и др.)
| Репликация вируса
|
|
|||
|
| |
| |
| |
| |
|
Продуктивная макрофагальная инфекция |
Продуктивная Т4-клеточная инфекция |
Поликлоналная активация В-клеток |
Снижение функции CD8-клеток |
|
|
|||
|
| |
| |
| |
| |
|
Инфицирование мозга: |
В т о р и ч н ы й иммунодефицит: |
||
|
| |
|
|
|
|
|
1. Инфекционный
синдром |
||
|
Нейро-психо-СПИД |
3. Аутоиммунный
синдром |
||
|
|
|||
|
Оппортунистическая инфекция: |
Саркома Капоши : |
Лимфома: |
|
|
1 место - легкие |
- кожа -92% |
- 1/3 - нервная система |
|
|
2 место - желудочно-кишечный тракт |
- прочая локализация - 8% |
- 1/4 - желудочно-кишечный тракт |
|
|
3 место - кожа и слизистые оболочки |
|
- 1/4 - костный мозг |
|
|
4 генерализованный процесс |
|
|
|
Таким образом, поражение иммунной системы при ВИЧ-инфекции носит системный характер, проявляясь глубокой супрессией Т- и В-звеньев клеточного иммунитета. В ходе развития ВИЧ-инфекции происходят закономерные изменения гиперчувствительности немедленного и замедленного типа, гуморального иммунитета и факторов неспецифической защиты, функциональной активности лимфоцитов и моноцитов/макрофагов. Нарастает уровень сывороточных иммуноглобулинов, циркулирующих иммунных комплексов, продуктов катаболизма клеточных рецепторов (Р-белки). происходят характерные изменения нуклеиновых кислот иммунокометентных клеток и активности в них ферментов основных обменных циклов. Наряду с дефицитом CD4+ лимфоцитов в динамике болезни нарастает функциональная недостаточность СD8+ лимфоцитов, NK-клеток, нейтрофилов. Нарушение иммунного статуса клинически проявляется инфекционным, аллергическим, аутоиммунным и лимфопролиферативным синдромами иммунологической недостаточности, синдромом, свойственным болезни иммунных комплексов. Все это определяет в целом клинику ВИЧ-инфекции.
Правда, имеется и
другое мнение о патогенезе ВИЧ-инфекции. Так, В. Колядин (1998) полагает, что
супрессия неспецифической защиты, а не дефицит Т-клеточного иммунитета
определяют формирование оппортунистической инфекции. Последняя ведет к дефициту
Т-клеточного звена иммунитета, а именно снижению количества Т-хелперов (CD4+
клетки). Отсюда, по мнению автора, дефицит CD4+ клеток - не причина, а следствие
развития оппортунистической инфекции. При всей привлекательности идеи автора
имеются и возражения. Как известно, прослеживается определенная связь между
характером иммунодефицита и видом возбудителя оппортунистической инфекции: c
клеточным связаны вирусная, грибковая инфекция, с гуморальныым - бактериальная.
У больных ВИЧ/СПИД основные виды суперинфекций представлены заболеваниями,
обусловленными вирусами, простейшими и грибами.